Aminolevulinic acid synthase
| 5-aminolevulinate synthase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 2.3.1.37 | ||||||||
| CAS number | 9037-14-3 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
ALA synthase (EC 2.3.1.37), or ALAS, catalyzes the synthesis of D-Aminolevulinic acid (ALA) the first common precursor in the biosynthesis of all tetrapyrroles such as hemes, cobalamins and chlorophylls.[1] The enzyme is expressed in all non-plant eukaryotes and the α-class of proteobacteria. Other organisms produce ALA through a three enzyme pathway known as the Shemin pathway. ALA is synthesized through the condensation of glycine and succinyl-CoA. In humans, transcription of ALA synthase is tightly controlled by the presence of Fe2+-binding elements, to prevent accumulation of porphyrin intermediates in the absence of iron. There are two forms of ALA synthase in the body. One form is expressed in red blood cell precursor cells (ALAS2), whereas the other (ALAS1) is ubiquitously expressed throughout the body. The red blood cell form is coded by a gene on chromosome x, whereas the other form is coded by a gene on chromosome 3. The disease X-linked sideroblastic anemia is caused by mutations in the ALA synthase gene on chromosome X, whereas no diseases are known to be caused by mutations in the other gene. Gain of function mutations in the erythroid specific ALA synthase gene have been shown recently to cause a previously unknown form of porphyria known as X-linked-dominant protoporphyria.
Enzyme Structure and Properties
PLP-dependent enzymes are prevalent because they are needed to transform amino acids into other resources.[1] ALAS is a homodimer with similarly sized sub units and the active sites consisting of amino acid side chains such as arginine, threonine, and lysine exist at a subunity interface.[1] The protein when extracted from R. spheroids contains 1600-folds and weighs about 80,000 daltons.[2] Enzymatic activity varies for different sources of the enzyme.[2]
Reaction Mechanism
The active sites of ALAS utilize three key amino acid side chains: Arg-85 and Thr-430 and Lys-313. Although these three amino acids have been identified to allow this reaction to proceed, they would be inactive without the addition of cofactor pyridoxal 5’-phosphate (PLP) whose role in this synthesis is detailed in the image above. Before the reaction can begin, the PLP cofactor binds to the lysine side chain to form a Schiff base that promotes attack by glycine substrate.[3][4][5][6] Lysine acts as a general base during this mechanism,.[1][7] In the detailed reaction mechanism, the hydronium atoms that are added in come from a variety of residues in that offer hydrogen bonds to facilitate ALA synthesis.[1] ALA synthase removes the carboxyl group from glycine and the CoA from the succinyl-CoA by means of its prosthetic group pyridoxal phosphate (a vitamin b6 derivative), forming δ-aminolevulinic acid (dALA), so called because the amino group is on the fourth carbon atom in the molecule. This reaction mechanism is particularly unique relative to other enzymes that use the PLP cofactor because Glycine is initially deprotonated by a highly conserved active site lysine, leading to condensation with succinyl-CoA and loss of CoA. Protonation of the carbonyl group of the intermediate by an active site histidine leads to loss of the carboxyl group. The last intermediate is finally reprotonated to produce ALA. Dissociation of ALA from the enzyme is the rate limiting step of the enzymatic reaction and was shown to be depended upon a slow conformational change of the enzyme. The function of pyridoxal phosphate is to facilitate the removal of hydrogen, by utilizing the electrophilic pyridinium ring as an electron sink.

The location of this enzyme in biological systems is indicative of the feedback that it may receive. ALA Synthase has been found in bacteria, yeast, avian and mammalian liver and blood cells and bone marrow. The location of this enzyme in animal cells is within the mitochondria.[2] Since the enzyme appears to be located near its source of succinyl-CoA and the end of the heme pathway indicates that the starting and end points of heme biosynthesis serves as feedback for ALA Synthase.[2] ALA synthase is also inhibited by hemin and glucose.[8]

Biological Function
ALAS1 and ALAS2 catalyze the first step in the process of heme synthesis. It is the first irreversible step and is also rate limiting. This means that the beginning of the formation of hemes is very intentional and subject to a variety of areas of feedback. For example, the two substrates, oxaloacetate and glycine, are highly produced by and utilized in other essential biological processes such as glycolysis and the TCA cycle. The image below illustrates the heme synthesis pathway and the role ALAS plays.
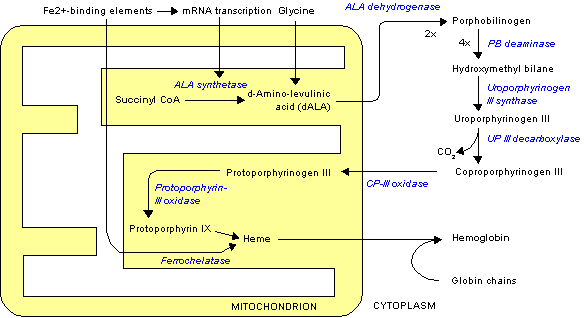
Disease Relevance
Aminolevulinic Acid Synthase Deficiency results in a lack of ability to create heme since its job is to catalyze the first step in the process. These deficiencies are often a result of genetic mutation that can result in a variety of diseases. One such disease is s-linked sideroblastic anemia which results in the appearance red blood cells in the bone marrow.[9] This disease is linked specifically with mutations in the genes that encode for ALAS2.[9]
References
- 1 2 3 4 5 Hunter, Gregory A.; Ferreira, Gloria C. (November 2011). "Molecular enzymology of 5-Aminolevulinate synthase, the gatekeeper of heme biosynthesis". Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1814 (11): 1467–1473. doi:10.1016/j.bbapap.2010.12.015.
- 1 2 3 4 Beale, S I (June 1978). "δ-Aminolevulinic Acid in Plants: Its Biosynthesis, Regulation, and Role in Plastid Development". Annual Review of Plant Physiology. 29 (1): 95–120. doi:10.1146/annurev.pp.29.060178.000523.
- ↑ "ALA synthase". flipper e nuvola. Turin University. Retrieved 10 March 2016.
- ↑ "5-Aminolevulinic acid synthase: mechanism, mutations and medicine.". Biochim Biophys Acta. 1647 (1-2): 361–6. Apr 2003. doi:10.1016/s1570-9639(03)00095-5. PMID 12686158.
- ↑ CHOI, H (July 2004). "Cloning, expression, and characterization of 5-aminolevulinic acid synthase from Rhodopseudomonas palustris KUGB306". FEMS Microbiology Letters. 236 (2): 175–181. doi:10.1016/j.femsle.2004.05.048.
- ↑ Ferreira, Gloria C.; Neame, Peter J.; Dailey, Harry A. (November 1993). "Heme biosynthesis in mammalian systems: Evidence of a schiff base linkage between the pyridoxal 5′-phosphate cofactor and a lysine residue in 5-aminolevulinate synthase". Protein Science. 2 (11): 1959–1965. doi:10.1002/pro.5560021117.
- ↑ Hunter, Gregory A.; Ferreira, Gloria C. (March 1999). "Lysine-313 of 5-Aminolevulinate Synthase Acts as a General Base during Formation of the Quinonoid Reaction Intermediates". Biochemistry. 38 (12): 3711–3718. doi:10.1021/bi982390w.
- ↑ Doss M, Sixel-Dietrich F, Verspohl F (1985). ""Glucose effect" and rate limiting function of uroporphyrinogen synthase on porphyrin metabolism in hepatocyte culture: relationship with human acute hepatic porphyrias.". J Clin Chem Clin Biochem. 23 (9): 505–13. doi:10.1515/cclm.1985.23.9.505. PMID 4067519.
- 1 2 Ajioka, Richard S.; Phillips, John D.; Kushner, James P. (July 2006). "Biosynthesis of heme in mammals". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1763 (7): 723–736. doi:10.1016/j.bbamcr.2006.05.005.
External links
- NIH
- Abu-Farha M, Niles J, Willmore W (2005). "Erythroid-specific 5-aminolevulinate synthase protein is stabilized by low oxygen and proteasomal inhibition". Biochem Cell Biol. 83 (5): 620–30. doi:10.1139/o05-045. PMID 16234850.
- Shemin, D; Rittenberg, D (1945). "The Utilization of Glycine for the Synthesis of a Porphyrin". J. Biol. Chem. 159: 567–8.
- SIDEROBLASTIC ANEMIAS -ALAS-2 defect disease