Dehydrogenative coupling of silanes
The dehydrogenative coupling of silanes is a powerful technique of the formation of Si-Si bonds in disilanes, oligosilanes and long, silicon-based polymers. These reactions are Wurtz-type coupling processes, and they generally must be catalyzed, as the rate-determining step is the kinetically unfavourable dissociation of hydrogen from silicon. Luckily, there are numerous transition metal complexes that can catalyze reactions between a wide variety of silanes, whether they be primary, secondary or tertiary.
Primary Silane
General Mechanism
Generally, the dehydrogenative coupling of primary silanes (as well as secondary and tertiary) proceeds via a Wurtz-type reaction, where two molecules of RSiH3 couple to form the corresponding dimer and eliminate one molecule of dihydrogen. Using an effective catalyst, this process can be extended to build long polysilanes.
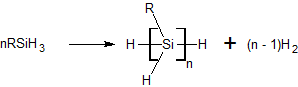
Catalyzed Reaction
Some of the catalysts that can be used in the dehydrogenative coupling of primary organosilanes are titanocene, zirconocene and their derivatives. While Wurtz-type coupling reactions are still the most common way to produce high molecular weight polysilanes, reactions using these titanocene and zirconocene catalysts and others like them have been improving and researchers are achieving higher molecular weight polysilanes as a result.[1]
Cp2Ti(OPh)2 (Cp = cyclopentadienyl) is a good catalyst for dehydrogenative coupling between primary silanes. This titanium catalyst is advantageous because it is highly active at room temperature, and in addition to silane polymerization, it can functionalize the ends via the hydrosilylation of an olefin, all in one step. For example, phenylsilane can be polymerized in the presence of vinyltrimethoxysilane, which produces a polysilane with a trimethoxysilane group at the end.[2]
While catalysts for dehydrogenative coupling reactions generally tend to be transition metal complexes, there has been some work done using magnesium oxide or calcium oxide for the reaction of phenylsilane and monosubstituted alkenes. This reaction is reported as being selective for only the dehydrogenative coupling and not the hydrosilylation reaction. In addition to high selectivity for dehydrogenative coupling, the reaction also has the advantage of being easy to separate through simple filtration of the reaction mixture to remove the solid magnesium oxide catalyst.[3]
The structures of the products of such reactions can generally be characterized with 1H, 13C and 29Si spectroscopy.
Wilkinson's Catalyst Reaction

Dehydrogenative coupling of primary silane using Wilkinson's catalyst is slow chemical reaction, but it does allow high chemoselectivity for the coupling reaction under appropriate conditions. Furthermore, no redistribution of product was observed by said reaction. A higher conversion percentage was observed for dehydrocoupling primary silanes under dynamic vacuum are consistent with the fact this reaction is a condensation reaction. Removal of H2 gas drives the reaction towards the coupled product. The sensitivity of primary dehydrogenative coupling of silane suggests the influence of subtle changes in the viscosity of the substrate. Experimentally, in an open system, a dramatic increase of coupling was observed when changing from n-dodecylsilane to a slightly less viscous n-octylsilane substrate.[4]
Relatively low volativity of the substrates allows us to efficiently remove hydrogen gas without removing any monosilane during reaction. However, it obstructs our ability to fully characterize olgiomeric products. Routine MS and GC-MS spectroscopy do not provide useful data for these substrates. ESI-MS can be used to analyze the catalytic reactions. Catalytical reactions involving labile phosphine ligand can be monitor via ESI-MS by the simple practice of tampering a fixed charged phosphine ligand.[5]
Spectroscopy
1H and 29Si NMR can sometimes be used to help identify and characterize the resulting products from these reactions. When using the 1H NMR spectra, it may be difficult to determine the details of the structure as often the peaks may show up as broad signals so it may only be useful in determining the integration of the signals as these are generally seen to match with what one would expect for the products. In the case of 29Si NMR, the spectra may also be poorly resolved but the use of DEPT can help in assigning silicone atoms of the product.[1]
Infrared spectroscopy may also be useful as it can indicate whether or not the product is a tertiary silane. The stretch for Si-H is seen at around 2100 and 910 cm−1. In the case of tertiary silanes however, the peak at 910 cm−1 is not seen. The shift or change in these peaks will be affected by the size of the polymer.[1]
Secondary silane
The polymerization of secondary silanes proceeds much the same way as it does for primary silanes. Many of the same catalysts can be used, though these reactions tend to be slower due to increased steric effects. Typically, dehydrogenative coupling of secondary silanes uses the Wurtz-type coupling reaction of halosilanes. The products are either cyclopolysilanes or linear polysilanes depending on the condition of the reaction. A likely intermediate of said reaction are halogen terminated olgiomers. However, the conversion of secondary silanes to olgiomers is less successful. The combination of the catalyst Cp2ZrCl2 and 2 equivalent nBuLi in toluene has successfully achieve polymerization of dehydrogenative coupling of secondary silane. The chain length ranges from 2 to 5 silanes and depends on the temperature, time, solvent and ratios of Si/Zr and Zr/Li.[6]
Tertiary Silanes
Due to steric factors, dehydrogenative coupling of tertiary silanes is a more difficult process, and many of the catalysts capable of coupling primary and secondary silanes will not work. However, disilane formation can be catalyzed with 4 mol % of CpFe(CO)2CH3 under photo-irradiation. This reaction follows a straightforward catalytic cycle. After the dissociation of one CO ligand, a tertiary silane R3SiH oxidatively adds to the iron centre, which induces the reductive elimination of methane. Then, the oxidative addition of a second silane unit occurs. From there, the disilane R3Si-SiR3 is reductively eliminated from the complex. Finally, after the oxidative addition of another silane unit and the reductive elimination of dihydrogen, the cycle is completed.[7]
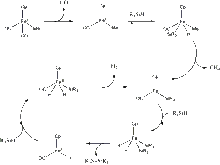
In addition, this same iron catalyst can be used to build polymers with a Si-R-Si backbone. When R = benzene, NH, or (C5H4)Fe(C5H4), this reaction works particularly well, and yields of over 95% are obtained.
B(C6F5)3 is another catalyst that can be used in the dehydrogenative coupling of tertiary silanes and has the useful characteristic of being selective for Si-H bonds over Si-Si bonds, leading to fewer branches and more linear polymers. This catalyst is particularly useful in reactions involving thiols and tertiary silanes or disilanes. Research into this reaction has found evidence that the rates of reactions catalyzed by this B(C6F5)3 complex are sensitive to steric factors as it is thought that the rate determining step in the dehydrogenative coupling using this catalyst involves the silane coming close to the catalyst, which is itself already quite bulky.[8]
As well as being coupled to each other, tertiary silanes can be coupled with carboxylic acids to form silyl esters. Ru3(CO)12/EtI is a good catalyst for this. Optimal reaction conditions are 1 - 8 mol % ruthenium catalyst, 4 mol % ethyl iodide in toluene under nitrogen, at 100 °C for 8 hours. If any larger amount of catalyst is used, or if the reaction runs longer or at a lower temperature, then much of the R3SiH will be converted into Et3SiH, which would result is a low yield of silyl ester. This reaction will work over a very large range of silanes and acids.[9]
The catalyst [Cu(PPh3)3Cl] can also be used to produce silyl esters in the dehydrogenative coupling of carboxylic acids and tertiary silanes. DMF or acetonitrile may be used for the solvent of the reaction and the reaction is done under a nitrogen atmosphere. The temperature of this reaction is important and for it to work requires that it be in the 80 – 120 °C range. At the same time, researchers have also investigated the use of triphenylphosphine as a catalyst for this reaction under similar conditions, but found that it requires a higher temperature and longer reaction time to generate the same type of yield.[10]
Tertiary silanes may also be dehydrogenatively coupled to aromatic rings with the use of the catalyst TpMe2Pt(Me)2H (TpMe2 = hydrido tris(3,5-dimethylpyrazolyl)borate). For example, this platinum catalyst can be used to react triethyl silane with benzene to produce phenyltriethylsilane, with the elimination of hydrogen gas. This is a terrific catalyst because it eliminates the need for a hydrogen acceptor, something which is normally required for the silation of a C-H bond. This reaction may also be done intramolecularly to produce five- or six-membered silicon-containing rings fused to a phenyl ring. In addition, tributylsilane can be converted into the corresponding cyclic organosilane via the same process. A drawback to this catalyst, however, is that it requires rather harsh reaction conditions (typically 200 °C for 24 hours for the intermolecular reaction, 48 – 72 hours for the intramolecular ones). It is also not particularly regioselective, so starting materials containing substituted benzene would result in a mixture of products.[11]

Polymerization of Silane
Traditionally, dehydrogenative coupling of silane involve Wurtz-Fittig coupling reaction. It involves the coupling of two halosilane and molten sodium. It can produce cyclic oligomers, polymers with high molecular weight, but it is limited by the number of tolerable functional groups, and control of product and low product yield.
Some methods used to produce polysilanes are polymerization of masked dislenes [12] , electroreduction of dichlorosilanes,[13] and dehydrocoupling of organosilanes using transition metal catalyst.
Organosilanes are organic derivatives of silane containing at least one carbon to silane bond. Dehydrocoupling of organosilane is an efficient method to produce polysilanes. One of the most successful routes to polymerize phenylsilane is via through group 4 metallocene derivatives. However, they provide polymers with low molecular weight. Nevertheless, the catalytic route allows the possibility to control the polymer stereochemistry as well as introducing new functionality to the group. Polysilanes are an interesting class of radiation sensitive material. They absorb and illuminate under ultraviolet light. This is attributed to the low lying σ- σ * transitions from delocalized electronic states of conjugated silicone atoms of the polymer. The substituent and molecular weight of the polymer influences the electronic properties of polysilane.[14]

Polymerization of para and meta substituted phenylsilanes commenced immediately with the addition of the catalyst. Ortho substituted polymers were failed to yield. Polymers were isolated as white/clear tacky materials and are soluble in most organic solvents. The polymers are shown to have no cross linking between the “X” group (X=H, alky, or halogen). Both steric and electronic effects influence the rate of dehydrocoupling of arylsilane. This coincides with the slow rate of the more steric hindered ortho substituted phenylsilane formation.[14]
Nickel catalyst reactions are few to come around with, and often come along with limitations. However, nickel catalysts are a great source of interest due to the cheap cost of nickel compared to other rare transition metals. Recently, [(dippe)Ni(µ-H)]2 nickel catalyst was found to dehydrogenative couple silane. The rate and extent of the polymerization of silane decreases as it proceeds from primary to secondary silane. The steric hindrance of secondary silane cause the polymerization to require a higher activation energy for silicon coupling at the metal center as the chain length increases. The dehydogenative coupling of organosilane catalyzed by [(dippe)Ni(µ-H)]2 can proceed through either ơ-bond metathesis or H2 extrusion followed by oxidation reaction. In an attempt to distinguish both mechanisms, the reaction of triethylsilane-d1 and [(dippe)Ni(µ-H)]2 was observed. A dehydrogenative coupling reaction of triethylsilane would not be catalyzed by the nickel. It would theoretically produce either H2 or HD gas via extrusion to support one mechanism. However, of Et3SiD and nickel hydrides was found and provide proof to support ơ-bond metathesis, but lack of dehydrogenative product of the silane does not allow either mechanism to be ruled out.[15]

References
- 1 2 3 Aitken, C.; Harrod, J. F.; Gill, U. S. (1987). "Structural studies of oligosilanes produced by catalytic dehydrogenative coupling of primary organosilanes". Can. J. Chem. 65: 1804–1809. doi:10.1139/v87-303.
- ↑ Garcia, J.; Meyer, D.; Guillaneux, D.; Moreau, J.; Wong, M. (2009). "Investigation of titanium-catalyzed dehydrogenate coupling and hydrosilation of phenylhydrogenosilanes in a one-pot process". J. Organ. Chem. (694): 2427–2433. doi:10.1016/j.organchem.2009.03.018.
- ↑ Itoh, M.; Mitsuzuka, M.; Utsumi, T.; Iwata, K.; Inoue, K. (1994). "Dehydrogenative coupling reactions between hydrosilanes and monosubstituted alkynes catalyzed by solid bases". Journal of Organometallic Chemistry. 476: C30–C31. doi:10.1016/0022-328X(94)87091-8.
- ↑ Rosenberg, Lisa; Kobus, Danielle N. (2003). "Dehydrogenative coupling of primary alkyl silanes using Wilkinson's catalyst". Journal of Organometallic Chemistry. 685 (1-2): 107–112. doi:10.1016/S0022-328X(03)00712-5.
- ↑ Jackson, Sarah M.; Chrisholm, Danielle M.; McIndoe, Scott J.; Rosenberg, Lisa (2011). "Using NMR and ESI-MS to probe the mechanism of silane dehydrocoupling Catalyzed by Wilkinson's Catalyst". European Journal of Inorganic Chemistry. 2011 (3): 327. doi:10.1002/ejic.201001165.
- ↑ Corey, J.Y.; Zhu, X.H.; Bedard, T.C.; Lange, L.D. (1991). "Catalytic Dehydrogenative Coupling of Secondary Silanes with Cp2MCl2". Organometallics. 10 (4): 924. doi:10.1021/om00050a024.
- 1 2 Itazaki, M.; Kensuke, U.; Nakazawa, H. (2009). "Iron-calalyzed dehydrogenative coupling of tertiary silanes". Angewandte Chemie. 48: 3313–3316. doi:10.1002/anie.200805112.
- ↑ Harrison, D. J.; Edwards, D. R.; McDonald, R.; Rosenberg, L. (2008). "Toward selective functionalisation of oligosilanes: borane-catalysed dehydrogenative coupling of silanes with thiols". Dalton Trans. 26: 3401–3411. doi:10.1039/b806270f.
- ↑ Liu, G.; Zhao, H. (2008). "Ru-catalyzed dehydrogenative coupling of carboxylic acids and silanes - a new method for the preparation of silyl esters". Beilstein Journal of Organic Chemistry. 4 (27). doi:10.3762/bjoc.4.27.
- ↑ Liu, G. -B.; Zhao, H. -Y.; Thiemann, T. (2007). "Two New Catalysts for the Dehydrogenative Coupling Reaction of Carboxylic Acids with Silanes - Convenient Methods for an Atom-Economical Preparation of Silyl Esters". Synthetic Communications. 37: 2717–2727. doi:10.1080/00397910701465669.
- 1 2 Tsukada, N.; Hartwig, J. F. (2005). "Intermolecular and intramolecular, platinum-catalyzed, acceptorless dehydrogenative coupling of hydrosilanes with aryl and aliphatic methyl C-H bonds". J.A.C.S. (127): 5022–5023. doi:10.1021/ja050612p.
- ↑ Sakomto, Kenkichi; Yoshida, Masaru; Sakurai, Hideki (1990). "Highly ordered high-molecular weight alternating polysilylene copolymer prepared by anionic polymerization of masked disilene". Macromolecules. 23 (20): 4494–4496. doi:10.1021/ma00222a031.
- ↑ Shono, T.; Ishifune, S.; Nishida, R. (1997). "Electroreductive synthesis of some functionalized polysilanes and related polymers". Tetrahedron Letters. 38 (36): 4607–4610. doi:10.1016/S0040-4039(97)00987-8.
- 1 2 Banovetz, John P.; Suzuki, Hiroshi; Waymouth, Robert M. (1993). "Dehydrogenative coupling of substituted phenylsilanes synthesis of poly[((trifluoromethyl)phenyl)silanes]". 12 (11): 4700–4703. doi:10.1021/om00035a070.
- 1 2 Smith, Erin E.; Du, Guodong; Fanwick, Phillipe E.; Abu-Omar, Mahdi M. (2010). "Dehydrocoupling of Organosilanes with a Dinuclear Nickel Hydride Catalyst and Isolation of a Nickel Silyl Complex". Organometallics. 29 (23): 6529. doi:10.1021/om100887v.