HH-suite
| Developer(s) | Johannes Söding, Michael Remmert, Andreas Biegert, Andreas Hauser, Markus Meier, Martin Steinegger |
|---|---|
| Stable release |
2.0.16
/ 18 February 2013 |
| Written in | C++ |
| Available in | English |
| Type | Bioinformatics tool |
| License | GPL v3 |
| Website | https://github.com/soedinglab/hh-suite |
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. These sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from their sequence.
Sequence searches in biology
Since the sequence of the human genome was determined in 2000, sequencing costs have dropped dramatically. Sequences for thousands of bacteria and hundreds of animals, plants, and fungi are filling the public sequence databases, but few of the functions of all the proteins encoded in these sequences are known. Even out of the approximately 20 000 human proteins, a large fraction of structures and functions remain unknown. To predict the function, structure, or other properties of a protein for which only its sequence of amino acids is known, the protein sequence is compared to the sequences of other proteins in public databases. If a protein with sufficiently similar sequence is found, the two proteins are likely to be evolutionarily related ("homologous"). In that case, they are likely to share similar structures and functions. Therefore, if a sufficiently similar protein with known functions and/or structure can be found by the sequence search, the unknown protein's functions, structure, and domain composition can be predicted. Such predictions greatly facilitate the determination of the function or structure by targeted validation experiments.
Description
The HH-suite HHsearch contains HHsearch [1] and HHblits [2] among other programs and utilities. HHsearch is among the most popular methods for the detection of remotely related sequences and for protein structure prediction, having been cited over 600 times in Google Scholar.[3] The HHsearch and HHblits programs owe their power to the fact that both the query and the database sequences are represented by multiple sequence alignments (MSAs). In these MSAs, the query or database sequence is written in a table together with homologous (related) sequences in such a way that each column contains homologous amino acid residues, that is, residues that have descended from the same residue in the ancestral sequence. The frequencies of amino acids in the columns of such an MSA can be interpreted as probabilities to observe an amino acid in a further homologous sequence at that position. To facilitate automatic scoring of potential sequences for their relatedness to the sequences in the MSA, the MSAs are succinctly described by profile hidden Markov models (HMMs). These are extensions of position-specific scoring matrices (PSSMs). The core algorithms for HMM-HMM alignment give HH-suite its name.
HHsearch takes as input a multiple sequence alignment or a profile hidden Markov Model (HMM) and searches a database of profile HMMs for homologous (related) proteins. HHsearch is often used for homology modeling, that is, to build a model of the structure of a query protein for which only the sequence is known: For that purpose, a database of proteins with known structures such as the protein data bank is searched for "template" proteins similar to the query protein. If such a template protein is found, the structure of the protein of interest can be predicted based on a pairwise sequence alignment of the query with the template protein sequence. In the CASP9 protein structure prediction competition in 2010, a fully automated version of HHpred based on HHsearch and HHblits was ranked best out of 81 servers in template-based structure prediction CASP9 TBM/FM.
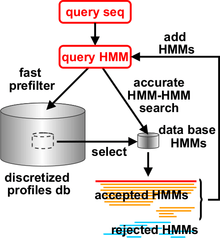
HHblits was added to the HH-suite in 2011. It can build high-quality multiple sequence alignments (MSAs) starting from a single query sequence or MSA. From the query, a profile HMM can be calculated. By using MSAs instead of single sequences, the sensitivity of sequence searches and the quality of the resulting sequence alignments can be improved dramatically. MSAs are also the starting point for a multitude of downstream computational methods, such as methods to predict the secondary and tertiary structure of proteins, to predict their molecular functions or cellular pathways, to predict the positions in their sequence or structure that contribute to enzymatic activity or ligand-binding, to predict evolutionarily conserved residues, disease-causing versus neutral mutations, the proteins' cellular localization and many more. This explains the importance to produce MSAs of the highest quality.
HHblits works similarly to PSI-BLAST, the most popular iterative sequence search method. HHblits generates a profile HMM from the query sequence and iteratively searches through a large database of profile HMMs, such as HH-suite's uniprot20 database. The uniprot20 database contains all public, high-quality protein sequences that are collected in the UniProt database. These sequences are clustered and aligned into multiple sequence alignments, from which the profile HMMs in uniprot20 are generated. Significantly similar sequences from the previous search are added to the query profile HMM for the next search iteration. Compared to PSI-BLAST and HMMER, HHblits is faster, up to twice as sensitive and produces more accurate alignments.[2] HHblits uses the same HMM-HMM alignment algorithms as HHsearch, but it employs a fast prefilter that reduces the number of database HMMs for which to perform the slow HMM-HMM comparison from tens of millions to a few thousands.
The HH-suite comes with a number of useful databases of profile HMMs that can be searched using HHblits and HHsearch, among them a clustered version of the UniProt database, HMMs for the protein data bank of protein structures, for the Pfam database of protein family alignments, the SCOP database of structural protein domains, and many more.
The HH-suite runs on most Linux and Unix distributions, including RedHat, Debian, Ubuntu, and Mac OS X. A Debian package is available.[4]
Overview of programs in HH-suite
In addition to HHsearch and HHblits, the HH-suite contains programs and perl scripts for format conversion, filtering of MSAs, generation of profile HMMs, the addition of secondary structure predictions to MSAs, the extraction of alignments from program output, and the generation of customized databases.
| hhblits | (Iteratively) search an HHblits database with a query sequence or MSA |
| hhsearch | Search an HHsearch database of HMMs with a query MSA or HMM |
| hhmake | Build an HMM from an input MSA |
| hhfilter | Filter an MSA by maximum sequence identity, coverage, and other criteria |
| hhalign | Calculate pairwise alignments, dot plots etc. for two HMMs/MSAs |
| reformat.pl | Reformat one or many MSAs |
| addss.pl | Add Psipred predicted secondary structure to an MSA or HHM file |
| hhmakemodel.pl | Generate MSAs or coarse 3D models from HHsearch or HHblits results |
| hhblitsdb.pl | Build HHblits database with prefiltering, packed MSA/HMM, and index files |
| multithread.pl | Run a command for many files in parallel using multiple threads |
| splitfasta.pl | Split a multiple-sequence FASTA file into multiple single-sequence files |
| renumberpdb.pl | Generate PDB file with indices renumbered to match input sequence indices |
References
- ↑ Söding J (2005). "Protein homology detection by HMM-HMM comparison". Bioinformatics. 21 (7): 951–960. doi:10.1093/bioinformatics/bti125. PMID 15531603.
- 1 2 Remmert M, Biegert A, Hauser A, Söding J (2011). "HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment.". Nat. Methods. 9 (2): 173–175. doi:10.1038/NMETH.1818. PMID 22198341.
- ↑ Number of citations to HHsearch on Google Scholar
- ↑ Debian hhsuite package
External links
- Soeding Lab at Max-Planck Institute in Göttingen - HH-suite developers
- HH-suite software and databases download from developers
- HHpred — free server at Max-Planck Institute in Tuebingen
- HHblits — free server at Max-Planck Institute in Tuebingen
- CASP website
- CASP9 template-based modeling results
- HH-suite debian package
- HH-suite ubuntu package
- HH-suite arch linux user repository