Hereditary multiple exostoses
| Hereditary multiple exostoses | |
|---|---|
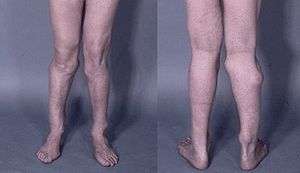 | |
| Photograph of the legs of a 26-year-old male showing multiple lumps leading to deformity. | |
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q78.6 |
| ICD-9-CM | 756.59 |
| OMIM | 133700 133701 |
| DiseasesDB | 33342 |
| Patient UK | Hereditary multiple exostoses |
| MeSH | D005097 |
Hereditary multiple exostoses (HME or MHE), also known as diaphyseal aclasis, is a rare medical condition in which multiple bony spurs or lumps (also known as exostoses, or osteochondromas) develop on the bones of a child. HME is synonymous with multiple hereditary exostoses and multiple osteochondromatosis, which is the preferred term used by the World Health Organization.
Presentation
HME can cause pain to people of all ages. To children, this can be especially painful. During exercise, it can cause a significant amount of pain. Exostoses may be visible to naked eye from outside. Multiple deformities, as mentioned above, can be present. The Exotoses appear to slow their rate of growth when they reach a certain, variable mass.
Pathophysiology
It is characterized by the growth of cartilage-capped benign bone tumours around areas of active bone growth, particularly the metaphysis of the long bones. Typically five or six exostoses are found in upper and lower limbs. Most common locations are:[1]
HME can lead to the shortening and bowing of bones; affected individuals often have a short stature. Depending on their location the exostoses can cause the following problems: pain or numbness from nerve compression, vascular compromise, inequality of limb length, irritation of tendon and muscle, Madelung's deformity[2] as well as a limited range of motion at the joints upon which they encroach. A person with HME has an increased risk of developing a rare form of bone cancer called chondrosarcoma as an adult.[2] Problems may be had in later life and these could include weak bones and nerve damage.[3][4][5] The reported rate of transformation ranges from as low as 0.57%[6] to as high as 8.3% of people with HME.[7]
Genetics
HME is an autosomal dominant hereditary disorder. This means that a patient with HME has a 50% chance of transmitting this disorder to his or her children. Most individuals with HME have a parent who also has the condition, however, approximately 10% -20% of individuals with HME have the condition as a result of a spontaneous mutation and are thus the first person in their family to be affected.
HME has thus far been linked with mutations in three genes.
- EXT1 which maps to chromosome 8q24.1[8]
- EXT2 which maps to 11p13[9]
- EXT3 which maps to the short arm of Chromosome 19 (though its exact location has yet to be precisely determined)[10]
Mutations in these genes typically lead to the synthesis of a truncated EXT protein which does not function normally. It is known that EXT proteins are important enzymes in the synthesis of heparan sulfate; however the exact mechanism by which altered synthesis of heparan sulfate that could lead to the abnormal bone growth associated with HME is unclear. It is thought that normal chondrocyte proliferation and differentiation may be affected, leading to abnormal bone growth.[11][12] Since the HME genes are involved in the synthesis of a glycan (heparan sulfate), HME may be considered a congenital disorder of glycosylation according to the new CDG nomenclature suggested in 2009.[13]
For individuals with HME who are considering starting a family, preimplantation genetic testing and prenatal diagnosis are available to determine if their unborn child has inherited the disease. HME has a 96% penetrance, which means that if the affected gene is indeed transmitted to a child, the child will have a 96% of actually manifesting the disease, and 4% chance of having the disease but never manifesting it. It should be noted that the 96% penetrance figure comes from one study.[6] Other studies have observed both incomplete and variable penetrance but without calculating the % penetrance, e.g.[14] In both the aforementioned studies the symptomless individuals carrying the faulty gene were predominantly female, leading to speculation that incomplete penetrance is more likely to be exhibited in females. Indeed, other work has shown that boys/men tend to have worse disease than females, as well as that the number of exostoses in affected members of the same family can vary greatly.[15] It is also possible for females to be severely affected.
Symptoms are more likely to be severe if the mutation is on the ext1 gene rather than ext2 or ext3; ext1 is also the most commonly affected gene in patients of this disorder.[15]
Possible connection to autism
Some parents of children with MHE have observed autism-like social problems in their children. To explore those observations more deeply, a 2012 study by the Sanford-Burnham Medical Research Institute used a mouse model of MHE to observe cognitive function. The findings indicated that the mutant mice endorsed three autistic characteristics: social impairment, impairments in ultrasonic vocalization, and repetitive behavior.[16]
Diagnosis
Diagnosis is mostly clinical and radiological. Technetium skeletal scintigrams are occasionally used to determine number of exostoses.[17]
Treatment
Surgical excision is performed when exostoses lead to growth disturbances or lead to disability. Knee osteotomies are associated with high incidence of peroneal nerve paralysis.[1]
Surgery, physical therapy and pain management are currently the only options available to HME patients, but success varies from patient to patient and many struggle with pain, fatigue and mobility problems throughout their lives. It is not uncommon for HME patients to undergo numerous surgical procedures throughout their lives to remove painful or deforming exostoses, correct limb length discrepancies or improve range of motion. Usually the treatment can be problematic. The osteochondromas can return in the same places and may be more painful.[4][18]
Epidemiology
HME is estimated to occur in 1 in 50,000 people.[1][5]
Additional images
.jpg) Multiple osteochondromas causing deformity of the forearm (shortening of the Radius with secondary bowing of the Ulna).
Multiple osteochondromas causing deformity of the forearm (shortening of the Radius with secondary bowing of the Ulna)..jpg) multiple osteochondromas at the pelvis
multiple osteochondromas at the pelvis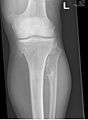 multiple osteochondromas around the knee
multiple osteochondromas around the knee CT of osteochondroma in MO
CT of osteochondroma in MO
References
- 1 2 3 Turek's orthopaedics principles and their application (6th ed.). Philadelphia: Lippincott Williams & Wilkins. 2005. p. 263. ISBN 9780781742986.
- 1 2 Davies, A. Mark; Pettersson, Holger (2002). Pettersson, Holger; Ostensen, Harald, eds. Radiography of the Musculoskeletal System (PDF). Geneva: World Health Organization. pp. 177, 189. ISBN 92-4-154555-0.
- ↑ CANNON JF (1954). "Hereditary multiple exostoses". American Journal of Human Genetics. 6 (4): 419–25. PMC 1716573
 . PMID 14349947.
. PMID 14349947. - 1 2 McBride WZ (September 1988). "Hereditary multiple exostoses". American Family Physician. 38 (3): 191–2. PMID 3046271.
- 1 2 Schmale GA, Conrad EU, Raskind WH (July 1994). "The natural history of hereditary multiple exostoses". The Journal of Bone and Joint Surgery. American Volume. 76 (7): 986–92. PMID 8027127.
- 1 2 Legeai-Mallet L, Munnich A, Maroteaux P, Le Merrer M (July 1997). "Incomplete penetrance and expressivity skewing in hereditary multiple exostoses". Clinical Genetics. 52 (1): 12–6. doi:10.1111/j.1399-0004.1997.tb02508.x. PMID 9272707.
- ↑ Kivioja A, Ervasti H, Kinnunen J, Kaitila I, Wolf M, Böhling T (March 2000). "Chondrosarcoma in a family with multiple hereditary exostoses". The Journal of Bone and Joint Surgery. British Volume. 82 (2): 261–6. PMID 10755438.
- ↑ Cook A, Raskind W, Blanton SH, Pauli RM, Gregg RG, Francomano CA, Puffenberger E, Conrad EU, Schmale G, Schellenberg G (1993). "Genetic heterogeneity in families with hereditary multiple exostoses". American Journal of Human Genetics. 53 (1): 71–9. PMC 1682231. PMID 8317501.
- ↑ Wu YQ, Heutink P, de Vries BB, Sandkuijl LA, van den Ouweland AM, Niermeijer MF, Galjaard H, Reyniers E, Willems PJ, Halley DJ (1994). "Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11". Human Molecular Genetics. 3 (1): 167–71. doi:10.1093/hmg/3.1.167. PMID 8162019.
- ↑ Le Merrer M, Legeai-Mallet L, Jeannin PM, Horsthemke B, Schinzel A, Plauchu H, Toutain A, Achard F, Munnich A, Maroteaux P (1994). "A gene for hereditary multiple exostoses maps to chromosome 19p". Human Molecular Genetics. 3 (5): 717–22. doi:10.1093/hmg/3.5.717. PMID 8081357.
- ↑ Zak BM, Crawford BE, Esko JD (2002). "Hereditary multiple exostoses and heparan sulfate polymerization". Biochimica et Biophysica Acta. 1573 (3): 346–55. doi:10.1016/S0304-4165(02)00402-6. PMID 12417417.
- ↑ Stieber JR, Dormans JP (2005). "Manifestations of hereditary multiple exostoses". The Journal of the American Academy of Orthopaedic Surgeons. 13 (2): 110–20. PMID 15850368.
- ↑ Jaeken J, Hennet T, Matthijs G, Freeze HH (2009). "CDG nomenclature: time for a change!". Biochim. Biophys. Acta. 1792 (9): 825–6. doi:10.1016/j.bbadis.2009.08.005. PMID 19765534.
- ↑ Faiyaz-Ul-Haque M, Ahmad W, Zaidi SH, et al. (August 2004). "Novel mutations in the EXT1 gene in two consanguineous families affected with multiple hereditary exostoses (familial osteochondromatosis)". Clinical Genetics. 66 (2): 144–51. doi:10.1111/j.1399-0004.2004.00275.x. PMID 15253765.
- 1 2 Porter DE, Lonie L, Fraser M, et al. (September 2004). "Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study". The Journal of Bone and Joint Surgery. British Volume. 86 (7): 1041–6. doi:10.1302/0301-620x.86b7.14815. PMID 15446535.
- ↑ Irie F, Badie-Mahdavi H, Yamaguchi Y (March 2012). "Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate". Proceedings of the National Academy of Sciences of the United States of America. 109 (13): 5052–6. doi:10.1073/pnas.1117881109. PMC 3323986. PMID 22411800.
- ↑ Grainger & Allison's diagnostic radiology: a textbook of medical imaging (5th ed.). Philadelphia: Churchill Livingstone/Elsevier. 2008. ISBN 978-0-443-10163-2.
- ↑ Morrissy R.T. and Weistein S.L. Lovell and Winter's pediatric orthopaedics, Volume 1, 5th ed., Lippincott Williams & Wilkins, Philadelphia, 2001; pp.274-275. ISBN 0-7817-2582-8
External links
- Hereditary Multiple Exostoses (MHE) Research Foundation's Website
- "Hereditary multiple exostoses". Medcyclopaedia. GE.
- GeneReviews: Hereditary Multiple Exostoses
- Information about Multiple Hereditary Exostoses (MHE)
- Hereditary Multiple Exostoses Support Group
- Images of Multiple Hereditary Exostoses (MHE) from Medical Image Database – MedPix