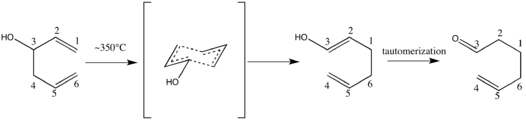
Oxy-Cope rearrangement
The oxy-Cope rearrangement is a variation of the Cope rearrangement in which 1,5-dien-3-ols are converted to unsaturated carbonyl compounds by a mechanism typical for such a [3,3]-sigmatropic rearrangement.[1][2] The reaction is highly general: a wide variety of precursors undergo the reorganization predictably and with ease, rendering it a highly useful synthetic tool.[3] Further, production of the required starting material is, on average, a relatively straightforward process. The modification was first proposed in 1964 by Berson and Jones, who coined the term. The driving force is the formation of a carbonyl via spontaneous keto-enol tautomerization.[4]
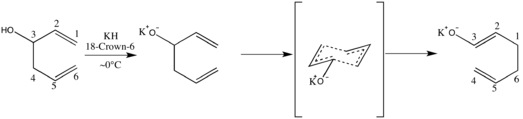
An additional modification of great significance was reported in 1975 by Evans and Golob, who added base to effect rate accelerations in the range of 1010-1017.[5] The authors termed the process an anionic oxy-Cope rearrangement.
The formation of an enolate renders the reaction irreversible in most cases,[3][4] its initial generation providing a force that greatly facilitates cleavage of the necessary carbon-carbon bonds.[6] Of further note is the great reduction in requisite temperatures and, in most contexts, the elimination of fragmentation as a competing reaction.
History
Sigmatropic rearrangements have been attributed great importance in the field of organic synthesis for their use as a readily exploitable stereoselective tool.[6] In an effort to demonstrate the versatility of the Cope rearrangement by demonstrating its tolerance of an alcohol situated at C-3 of a 1,5-diene, Berson and Jones heated a bicyclic diene alcohol in the gas phase to give cis-∆5,6-octalone in fair yield.[1]
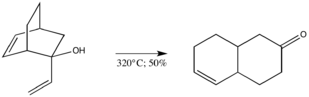
The modification is immensely appealing as a result of the two new disparately placed functional groups that lend themselves well to a variety of previously unavailable synthetic manipulations.
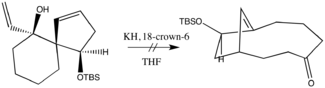
The next development occurred in 1975, when Evans and Golob reported tremendous anionic-assisted rate enhancements. Their use of potassium hydride in the cation’s corresponding crown ether quickly replaced thermal conditions as the default approach—for most applications. Indeed, in some cases, anionic assistance is intentionally forgone to accommodate for the production of overly sensitive enolate product. For example, in the following reaction only tar was obtained, a result that the authors attributed to the product’s ostensible intolerance to base.[7] The original oxy-Cope modification thus to this day occupies a relevant niche in synthetic chemistry.
Mechanism
Both the neutral and anionic variants of the oxy-Cope rearrangement may occur via either concerted or stepwise radical pathways, although the former mode is generally favored.[8][9] The preferred intermediate is characterized by a chair-like conformation.[10] It is expressly a result of this highly ordered transition state that chirality transfer is effected in a predictable manner.[4][10] Specifically, the geometric positioning of the double bonds in the most readily accessible transition state for a given reaction directly determines the stereochemical result of the reaction.[3] A boat transition state is disfavored, but typically rearrangements occur via this path to an appreciable extent as well. The result is the production of diastereomeric mixtures.
As demonstrated by the variance in resultant mixtures, sterically demanding substituents alter a substrate’s relative tendency to rearrange through its preferred transition state.[11]
Rearrangements for which a chair transition state is geometrically impossible can nonetheless occur. In fact, enolate formation provides enough of a driving force to overcome the energetic barrier associated with both dearomatization and the boat conformer.[12]
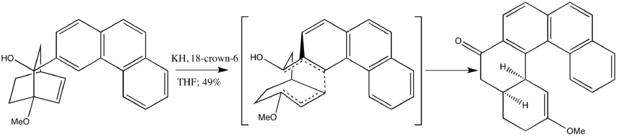
The concerted, synchronous pathways presented above generally predominate; it was calculated for anionic oxy-Cope processes that a divide of 17-34 kcal/mol favors heterolysis over homolysis.[13] Several factors may bridge this energetic gap, however, as evidenced in the work of Cha, Kim, and Cho.[10]

The large degree of strain and the presence of a methyl group’s bulk favored the (Z)- instead of the expected (E)-cyclooctenone isomer, suggesting that the intermediate was not formed synchronously. Only with fragmentation and subsequent isomerization steps could the observed product be rationalized.[10]
Interestingly, a study on the anionic oxy-Cope rearrangement carried out entirely in the gaseous phase reported that the rate enhancement stems not from solvent interactions, but from those within the structure itself.[14]
Rate Enhancement
In general, decreasing the stability of the oxy-Cope or anionic oxy-Cope substrate relative to that of the product results in increased rate of reaction by the principle of ground state destabilization. This desirable outcome is readily achieved in a variety of ways. In their seminal paper Evans and Golob reported on the importance of ionic interactions between metal and alkoxide, citing an increase in dissociative character as a cause for rate acceleration.[5] Their use of 15-crown-5 in conjunction with sodium hydride afforded a 1.27-fold rate enhancement over the course of a bicyclic diene alkoxide’s sigmatropic conversion to enolate product, while the same reaction with HMPT in 15-crown-5’s place did not appreciably affect the rate. Interestingly, the use of potassium hydride in conjunction with 18-crown-6 to achieve the same end afforded a 180-fold maximum rate acceleration. From the above results it was concluded that rate increases as counterions more poorly approximate point charges—and with the addition of counterion-sequestering species.
The inclusion of more polar solvents and catalytic quantities of phase transfer salts has also been demonstrated to exert the same rate-enhancing effect.[15]
Finally, the relief of ring strain over the course of a rearrangement will drive a reaction more forcibly to completion, thereby increasing its rate.
Scope
Because there exist multiple classes of natural products containing eight-membered rings, the syntheses of which having proved difficult, the anionic oxy-Cope rearrangement has been highlighted as a suitable alternative pathway. Its application here offers great stereochemical control, and its use is far more general than the relatively unsuccessful routes that had been employed before its development.[16]
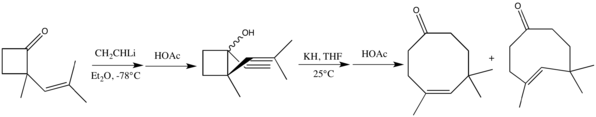
In spite of possible geometrical constraints, the required unsaturated substrates may contain triple bonds in place of either of the double bonds. Such an alkynol was effectively manipulated in the elegant synthesis of both poitediol and dactylol.[6] These interesting sigmatropic rearrangements can occur either with anionic assistance or under thermal conditions.[17]
Of particular interest is the application of the oxy-Cope to situations in which the immediate product reacts further in a predictable manner to furnish a desired final product. This goal was achieved in the synthesis of the cis-hydroazulenone below, in which the oxy-Cope intermediate was characterized by a stereoelectronic configuration amenable to remote SN displacement.[18]
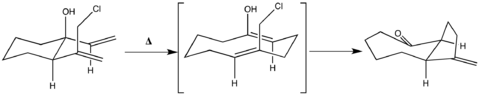
Further Considerations
Potassium hydroxide, a frequently utilized reagent for the anionic oxy-Cope rearrangement, is occasionally contaminated with trace impurities that have been suggested to destroy the dienolate intermediate, resulting in putative polymerization. Macdonald et. al, who documented the occurrence, prescribed pre-treatment with iodine to eliminate any potassium superoxide that may persist within a purchased batch of the material. This simple preparatory step, as they describe in their paper, effects dramatic improvement in both yield and reproducibility of results.[19]
Important side reactions include heterolytic cleavage, in which the homoallylic alcohol decomposes into a carbonyl and an allylic system.[20]
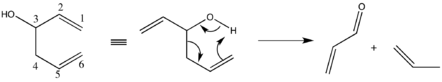
Suppression of this phenomenon is readily achievable by decreasing the ionic nature of the metal-alkoxide bond. Specifically, the use of more electronegative alkali metals or solvents less amenable to cation solvation generates the desired effect.[21] In keeping with the above discussion, the rate of reaction may be diminished but should not approach an unsatisfactory level.
References
- 1 2 Berson, Jerome A.; Jones, Maitland (1964). "A Synthesis of Ketones by the Thermal Isomerization of 3-Hydroxy-1,5-hexadienes. The Oxy-Cope Rearrangement". J. Am. Chem. Soc. 86 (22): 5019–5020. doi:10.1021/ja01076a067.
- ↑ Cope, Arthur C.; Hardy, Elizabeth M. (1940). "The Introduction of Substituted Vinyl Groups. V. A Rearrangement Involving the Migration of an Allyl Group in a Three-Carbon System". J. Am. Chem. Soc. 62 (2): 441–444. doi:10.1021/ja01859a055.
- 1 2 3 Paquette, Leo A. (1997). "Recent Applications of Anionic Oxy-Cope Rearrangements". Tetrahedron. 53 (41): 13971–14020. doi:10.1016/S0040-4020(97)00679-0.
- 1 2 3 Kürti, Lázló; Czakó, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis. Burlington: Elsevier Inc. p. 325.
- 1 2 Evans, D. A.; Golob, A. M. (1975). "[3,3]Sigmatropic Rearrangements of 1,5-Diene Alkoxides. The Powerful Accelerating Effects of the Alkoxide Substituent". J. Am. Chem. Soc. 97 (16): 4765–4766. doi:10.1021/ja00849a054.
- 1 2 3 Wilson, Stephen R. (1993). "Anion-Assisted Sigmatropic Rearrangements". Org. React. 43 (2): 93–250. doi:10.1002/0471264180.or043.02.
- ↑ Paquette, Leo A.; Ladouceur, Gaetan (1989). "Synthetic Studies Targeted at the Cytotoxic 8,9-Seco-ent-kaurene Diterpenes. Concise Complementary Stereocontrolled Construction of the Bridgehead Olefin Core". J. Org. Chem. 54 (18): 4278–4279. doi:10.1021/jo00279a010.
- ↑ Evans, D. A.; D. J., Baillargeon (1978). "Alkoxide Substituent Effects on Carbon—Carbon Bond Homolysis". Tetrahedron Letters. 19 (36): 3319–3322. doi:10.1016/S0040-4039(01)85627-6.
- ↑ Paquette, Leo A.; Pierre, Francis; Cottrell, Charles E. (1987). "Anionic Rearrangements of syn- and anti-7-Cyclopentenyl-7-hydroxynorbornenes. The Case for Sequential Ring Cleavage and Intramolecular Michael Addition". J. Am. Chem. Soc. 109 (19): 5731–5740. doi:10.1021/ja00253a027.
- 1 2 3 4 Maurin, Philippe; Kim, Se-Ho; Cho, Sung Yun; Cha, Jin Kun (2003). "On the Mechanism of the Anionic Oxy-Cope Rearrangement of trans-1,2-Dialkenylcyclobutanols". Angew. Chem. 42 (41): 5044–5047. doi:10.1002/anie.200350988.
- ↑ Evans, D. A.; Nelson, John V. (1980). "Stereochemical Study of the [3,3] Sigmatropic Rearrangement of 1,5-Diene-3-alkoxides. Application to the Stereoselective Synthesis of (.+-.)-Juvabione". J. Am. Chem. Soc. 102 (2): 774–782. doi:10.1021/ja00522a056.
- ↑ Ogawa, Yasushi; Ueno, Tetsuya; Karikomi, Michinori; Seki, Katsura; Haga, Kazuo; Uyehara, Tadao (2001). "Synthesis of 2-Acetoxy[5]helicene by Sequential Double Aromatic Oxy-Cope Rearrangement". Tetrahedron Lett. 43: 7827–7829. doi:10.1016/s0040-4039(02)01611-8.
- ↑ Evans, D.A.; Baillargeon, D.J. (1978). "Intrinsic Fragmentation Modes of Primary Alkoxides". Tetrahedron Lett. 19 (36): 3315–3318. doi:10.1016/S0040-4039(01)85626-4.
- ↑ Baldwin, John E.; Black, Kersey A. (1984). "Complete Kinetic Analysis of Thermal Stereomutations among the Eight 2,3-Dideuterio-2-(methoxymethyl)spiro[cyclopropane-1,1'-indenes]". J. Am. Chem. Soc. 106 (4): 1029–1040. doi:10.1021/ja00316a036.
- ↑ Georges, Michael; Tam, Tim F.; Fraser-Reid, Bert (1985). "Controlled Access to Furanose Precursors Related to Sesquiterpene Lactones. 1". J. Org. Chem. 50 (26): 5747–5753. doi:10.1021/jo00350a062.
- ↑ Gadwood, Robert C.; Lett, Renee M. (1982). "Preparation and Rearrangement of 1,2-Dialkenylcyclobutanols. A Useful Method for Synthesis of Substituted Cyclooctenones". J. Org. Chem. 47 (12): 2268–2275. doi:10.1021/jo00133a007.
- ↑ Viola, Alfred; MacMillan, John H. (1970). "Vapor Phase Acetylenic Oxy-Cope Reaction of 5-Hexen-1-yn-3-ol. The Chemistry of an Allenol Intermediate". J. Am. Chem. Soc. 92 (8): 2404–2410. doi:10.1021/ja00711a034.
- ↑ Sworin, Michael; Lin, Ko Chung (1987). "Cyclopentanoid Synthesis via the Intramolecular Trapping of Oxy-Cope Intermediates. Stereocontrolled Synthesis of the cis- and trans-Hydroazulene Skeleton". J. Org. Chem. 52 (25): 5640–5642. doi:10.1021/jo00234a029.
- ↑ Macdonald, Timothy L.; Natalie, Kenneth J.; Prasad, Girija; Sawyer, J. Scott (1986). "Chemically Modified Potassium Hydride. Significant Improvement in Yields in Some Oxy-Cope Rearrangements". J. Org. Chem. 51 (7): 1124–1126. doi:10.1021/jo00357a035.
- ↑ Snowden, Roger L.; Muller, Bernard L.; Schulte-Elte, Karl H. (1981). "Fragmentation of Homoallylic Alkoxides. Synthesis of Propenyl and 2-Methylpropenyl Ketones from Carboxylic Esters". Tetrahedron Lett. 23 (3): 335–338. doi:10.1016/S0040-4039(00)86824-0.
- ↑ Evans, D. A.; Baillargeon, David J.; Nelson, John V. (1978). "A General Approach to the Synthesis of 1,6-Dicarbonyl Substrates. New Applications of Base-Accelerated Oxy-Cope Rearrangements". J. Am. Chem. Soc. 100 (7): 2242–2244. doi:10.1021/ja00475a051.