Reactions of alkenyl- and alkynylaluminium compounds
Reactions of alkenyl- and alkynylaluminium compounds involve the transfer of a nucleophilic alkenyl or alkynyl group attached to aluminium to an electrophilic atom. Stereospecific hydroalumination, carboalumination, and terminal alkyne metalation are useful methods for generation of the necessary alkenyl- and alkynylalanes.[1]
Introduction
Aluminium, like its congener boron, is less electronegative than carbon (Al, 1.61; C, 2.55); thus, aluminium-bound carbons in organoalanes possess partial negative charge and are nucleophilic as a result. Generally, however, organoalanes are not nucleophilic enough to transfer an organic group on their own (the exception being when carbonyl and enone acceptors are used, due to the high oxophilicity of aluminium[2]). In most cases, nucleophilic activation of organoalanes is necessary for group transfer to take place. Like organoboranes, organoalanes possess an empty p orbital on the aluminium center that can receive electron density from an added nucleophile. The resulting negatively charged aluminate is much more nucleophilic than the neutral alane.[3]
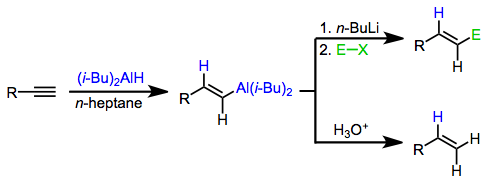
This concept has been applied to methods for the synthesis of organic compounds from alkenyl- and alkynylalanes. The most notable applications are methods for the stereospecific synthesis of olefins. Alkenylalanes, which are easily synthesized with complete stereocontrol through alkyne hydroalumination, transfer the alkenyl unit to a variety of electrophiles. Alkynylalanes are less commonly used because alkali metal acetylides can be used for many of the same transformations as alkynylalanes. However, alkynylalanes are useful for the coupling of tertiary halides and alkynes (a reaction difficult to effect with alkali metal alkynes)[4] and for conjugate addition[5] and epoxide opening reactions.[6]
(1)
Mechanism and Stereochemistry
Hydroalumination of Alkynes
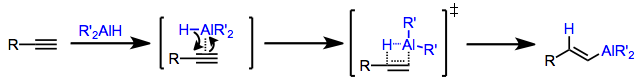
Hydroalumination of alkynes may be either stereospecifically cis or trans depending on the conditions employed. When a dialkylalane such as di(isobutyl)aluminium hydride (DIBAL-H) is used, the hydrogen and aluminium delivered from the reagent end up cis in the resulting alkenylalane.[7] This stereospecificity can be explained by invoking a concerted addition of the H–Al bond across the triple bond. In the transition state, partial positive charge builds up on the carbon forming a bond to hydrogen; thus, the carbon better able to stabilize a positive charge becomes attached to hydrogen in the product alkenylalane.[8] Hydroaluminations of terminal alkynes typically produce terminal alkenylalanes as a result. Selectivity in hydroaluminations of internal alkynes is typically low, unless an electronic bias exists in the substrate (such as a phenyl ring in conjugation with the alkyne).[9]
(2)
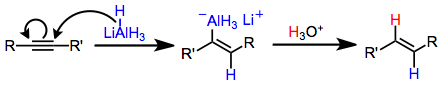
Stereospecific trans hydroalumination is possible through the use of lithium aluminium hydride. The mechanism of this transformation involves the addition of hydride to the carbon less able to stabilize the developing negative charge (viz., in the β position to an electron-withdrawing group).[10] Coordination of aluminium to the resulting trans vinyl carbanion leads to the observed trans configuration of the product.[11]
(3)
Reactions of Alkenyl- and Alkynylalanes and Aluminates
Neutral alanes are not nucleophilic enough to deliver organic groups to electrophilic substrates. However, upon activation by a nucleophile, the resulting aluminates are highly nucleophilic and add to electrophiles with retention of configuration at the migration carbon. Thus, stereospecific hydroalumination followed by nucleophilic attack provides a method for the stereospecific synthesis of olefins from alkynes.[3]
(4)

Scope and Limitations
Because unsaturated alanes are oxygen- and moisture-sensitive, they are most often prepared for immediate use without isolation. However, the method of preparation determines the configuration of the intermediate unsaturated alane, which is directly related to the configuration of the product (transfer of the alkenyl group occurs with retention of configuration). Thus, an understanding of available hydroalumination methods is important for the study of reactions of unsaturated alanes. This second describes the most common methods of hydroalumination, and subsequent chemical reactions that the resulting alkenylalanes may undergo.
Preparation of Alkenylalanes
Stereospecific cis-hydroalumination is possible through the use of dialkylalanes. The most common reagent used for this purpose is di(isobutyl)aluminium hydride (DIBAL-H). Analogous to hydroboration reactions with R2BH, hydroalumination with R2AlH leads to the attachment of aluminium at the carbon less able to stabilize developing positive charge (anti-Markovnikov selectivity).[12] Metalation of terminal alkynes is a significant side reaction that occurs under these conditions. If metalation is desired, tertiary amine complexes of DIBAL-H are useful.[13]
(5)
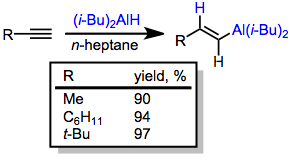
The use of silyl acetylenes avoids the problem of competitive metalation of terminal alkenes. The stereoselectivity of hydroalumination can be altered through a change in solvent: tertiary amine solvents provide the trans alkenylalane and hydrocarbon solvents provide the cis isomer.[14]
(6)

Lithium aluminium hydride hydroaluminates alkynes to afford trans alkenylalanes. In equation (7) hydride adds to the terminal carbon, which places the developing negative charge next to the stabilizing phenyl substituent.[10]
(7)

Reactions of Alkenylalanes
Alkenyl- and alkynylaluminates are most commonly generated through the addition of n-butyllithium to the alkenylalane. The alkenyl and alkynyl groups, which are better able to stabilize negative charge, are transferred in preference to the alkyl group. When these intermediates react with alkyl halides, functionalized olefins are produced.[15]
(8)

Treatment of alkenylaluminates with halogen electrophiles such as N-bromosuccinimide (NBS) and iodine leads to the formation of halogenated olefins.[16] These products are useful for cross-coupling reactions.
(9)

Zirconium-catalyzed carboalumination of alkynes by trimethylalane is a convenient method for accessing substituted alkenylalanes stereoselectively.[17] Upon exposure to aldehydes and ketones, alkenylalanes form secondary or tertiary allylic alcohols. Formaldehyde is a useful reagent in this context for the introduction of a hydroxymethyl unit.[18]
(10)
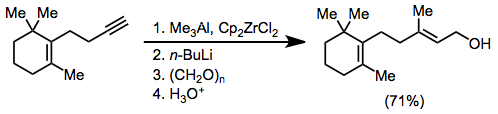
Alkynylalanes are primarily used in place of the corresponding alkali metal acetylides when the latter reagents are ineffective. The coupling of an acetylide and tertiary alkyl halide is an example of a reaction that cannot be accomplished with alkali metal acetylides, which displace halides in an SN2 fashion. The corresponding alkynylalanes are able to couple to tertiary halides via an SN1-like mechanism.[4]
(11)

Alkynyl- and alkenylalanes add in a conjugate fashion to enones in the s-cis conformation without nucleophilic activation. Enones locked in an s-trans conformation, such as cyclohexenone, are unreactive. The coordination of oxygen to aluminium is believed to be necessary for this reaction.[19]
(12)

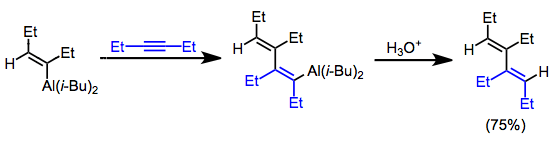
When alkynes and dialkylalanes are combined in a 2:1 ratio, 1,3-dienes result. The aluminium-carbon bond of the initially formed alkenylalane adds across a second molecule of alkyne, forming a conjugated dienylalane. Protonolysis provides the metal-free diene product.[20]
(13)
Alkenyl- and alkynylalanes undergo transmetalation to a variety of metals, including boron,[21] zirconium,[21] and mercury.[22]
(14)

Experimental Conditions and Procedure
Typical Conditions
Organoaluminium compounds are extremely pyrophoric and should be handled only under inert atmosphere. Dialkylaluminium hydrides and lithium aluminium hydride are both available commercially; the former is available either neat or in solutions that may be standardized using known procedures.[23] Solvents and other reagents should be scrupulously dried. Workup of these reactions should employ extreme pH's (10% hydrochloric acid or 6 N sodium hydroxide), as moderate pH encourages the formation of gelatinous aluminium hydroxide, which renders product separation difficult.
Example Procedure
(15)

To 2.76 g (25.0 mmol) of 1-octyne was added 25.0 mL of a 1.07 M solution of diisobutylaluminium hydride (26.8 mmol) in n-hexane while the temperature was maintained at 25–30° by means of a water bath. The solution was stirred at room temperature for 30 minutes and then was heated at 50° for 4 hours. The resultant alkenylalane was cooled to –30°, diluted with 15 mL of dry ether, and treated with 5.35 g (30.1 mmol) of N-bromosuccinimide while keeping the temperature below –15°. The reaction mixture was gradually warmed to room temperature and stirred for 1 hour before being poured slowly into a mixture of 6 N hydrochloric acid (50 mL), n-pentane (10 mL), and ice (10 g). The layers were separated, and the aqueous phase was extracted with pentane. The combined organic extract was washed successively with 1 N sodium hydroxide, 10% sodium sulfite, and saturated aqueous sodium chloride and then was treated with a few crystals of BHT to inhibit isomerization of the alkenyl bromide. After drying over magnesium sulfate, distillation afforded 3.72 g (78%) of (E)-1-bromo-1-octene, bp 67° (5 mm), nD26 1.4617. This compound, which contained 4% of 1-bromo-1-octyne, was stored over a few crystals of BHT. 1H NMR (CDCl3): δ 0.92 (m, 3 H), 1.1-1.7 (m, 8 H), 1.90-2.34 (m, 2 H), 5.85-6.30 (m, 2 H).
References
- ↑ Zweifel, G.; Miller, J. A. Org. React. 1984, 32, 375. doi:10.1002/0471264180.or032.02
- ↑ Collins, P. W.; Dajani, E. Z.; Bruhn, M. S.; Brown, C. H.; Palmer, J. H.; Pappo, R. Tetrahedron Lett. 1975, 4217.
- 1 2 Negishi, E.-i. J. Organometal. Chem. Lib. 1976, 1, 93.
- 1 2 Negishi, E.-i.; Baba, S. J. Am. Chem. Soc. 1975, 97, 7385.
- ↑ Hooz, J.; Layton, R. B. J. Am. Chem. Soc. 1971, 93, 7320.
- ↑ Danishefsky, S.; Kitahara, T.; Tsai, M.; Dynak, J. J. Org. Chem. 1976, 41, 1669.
- ↑ Wilke, G.; Müller, H. Chem. Ber. 1956, 89, 444.
- ↑ Bundens, J. W.; Francl, M. M. Organometallics 1993, 12, 1608.
- ↑ Eisch, J. J.; Gopal, H.; Rhee, S.-G. J. Org. Chem. 1975, 40, 2064.
- 1 2 Slaugh, L. H. Tetrahedron 1966, 22, 1741.
- ↑ House, H. O. Modern Synthetic Reactions, 2nd ed., Benjamin/Cummings, Menlo Park, CA, 1972.
- ↑ Eisch, J. J.; Kaska, W. C. J. Am. Chem. Soc. 1963, 85, 2165.
- ↑ Binger, P. Angew. Chem. Int. Ed. Engl. 1963, 2, 686.
- ↑ Eisch, J. J.; Foxton, M W. J. Org. Chem. 1971, 36, 3520.
- ↑ Zweifel, G.; Lynd, R. A. Synthesis 1976, 816.
- ↑ Palei, B. A.; Gavrilenko, V. V.; Zakharkin, L. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1969, 2760 [C.A., 72, 79143s (1970)].
- ↑ Yoshida, T.; Negishi, E.-i. J. Am. Chem. Soc. 1981, 103, 4985.
- ↑ Negishi, E.-i.; King, A. O.; Klima, W.; Patterson, W.; Silveira, Jr., A. J. Org. Chem. 1980, 45, 2526.
- ↑ Hooz, J.; Layton, R. B. Can. J. Chem. 1973, 51, 2098.
- ↑ Zweifel, G.; Snow, J. T.; Whitney, C. C. J. Am. Chem. Soc. 1968, 90, 7139.
- 1 2 Negishi, E.-i.; Boardman, L. D. Tetrahedron Lett. 1982, 3327.
- ↑ Negishi, E.-i.; Jadhav, K. P.; Daotien, N. Tetrahedron Lett. 1982, 2085.
- ↑ Brown, H. C. Organic Syntheses via Boranes, Wiley, New York, 1975.