Methylglyoxal synthase
| methylglyoxal synthase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 4.2.3.3 | ||||||||
| CAS number | 37279-01-9 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / EGO | ||||||||
| |||||||||
In enzymology, a methylglyoxal synthase (EC 4.2.3.3) is a enzyme that catalyzes the chemical reaction
- dihydroxyacetone phosphate methylglyoxal + phosphate

Hence, this enzyme has one substrate, DHAP, and two products, methylglyoxal and phosphate. Attempts to observe reversibility of this reaction have been unsuccessful.[1]
This enzyme belongs to the family of lyases, specifically those carbon-oxygen lyases acting on phosphates. The systematic name of this enzyme class is glycerone-phosphate phosphate-lyase (methylglyoxal-forming). Other names in common use include methylglyoxal synthetase, and glycerone-phosphate phospho-lyase. This enzyme participates in pyruvate metabolism and is constitutively expressed.[1]
Structural Studies
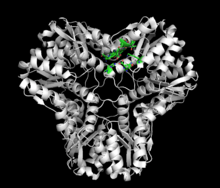
As of late 2007, 7 structures have been solved for this class of enzymes, with PDB accession codes 1B93, 1EGH, 1IK4, 1S89, 1S8A, 1VMD, and 1WO8.
Methylglyoxal synthase (MGS) is a 152-amino acid homohexamer that has a molecular weight of approximately 67,000 kD.[2][3][4] The total solvent-accessible surface area of the MGS homohexamer is 18,510 square Angstroms, roughly 40% of the total possible surface area if the subunits were separated.[3] Each monomer consists of five alpha helices surrounding five beta sheets. Of these, two antiparallel beta sheets and one alpha helix are located in a subdomain where the N-terminus and C-terminus are in close juxtaposition.[3] The homohexamer exhibits a threefold axis perpendicular to a twofold axis. Within the wide V-groove, there are twelve hydrogen bonds and six salt bridges between the monomers in the presence of phosphate binding. In the absence of phosphate binding, ten hydrogen bonds and two salt bridges hold the monomers together. At the peak interfaces, ten hydrogen bonds and no salt bridges connect the monomers regardless of phosphate binding.[3]
The MGS homohexamer is slightly asymmetrical. All three monomers within the asymmetrical region contain a formate molecule within their respective actives sites. Only one of the monomers within the asymmetrical region is additionally bound to a phosphate.[3]
The active site contains many conserved residues for function (Asp, His, Thr) and structure (Gly, Pro). Inorganic phosphate interacts with Lys23, Thr45, Thr47, Thr48, and Gly66. Formate interacts with His19, His98, and Asp71. The active site is exposed to the solvent via a perpendicular channel that consists of Arg150, Tyr146, Asp20, Pro67, His98, and His19.[3]
Although mechanistically similar to triosephosphate isomerase (TIM), MGS contains widely dissimilar protein folding that prevents structural alignment with TIM which suggests convergent evolution of their chemical reactions. However, Asp71 in MGS may act similarly to the Glu165, the catalytic base in TIM. Additionally, His19 and His98 may perform the role of the electrophilic catalyst similar to His95 in TIM. CheB methylesterase has the highest structural similarity with MGS.[3]
Mechanism
Methylglyoxal synthase is highly specific for DHAP with Km 0.47mM at its optimal pH of 7.5.[2][5] Contrary to early reports, the purified enzyme does not react with other glycolytic metabolites such as glyceraldehyde-3-phosphate or fructose 1,6-diphosphate.[2][6] The mechanism of MGS is similar to that of TIM; both enzymes react with dihydroxyacetone phosphate to form an ene-diol phosphate intermediate as the first step of their reaction pathways.[3] However, the second step involves the elimination of phosphate to form methylglyoxal instead of reprotonation to form glyceraldehyde-3-phosphate.[3] The overall reaction is characterized as an intramolecular oxidation-reduction followed by a dephosphorylation. The C-3 of DHAP is oxidized to an aldehyde, while C-1, which bears the phosphate ester, is dephosphorylated and reduced to a methyl group.[7] MGS does not require the use of metal ions or a Schiff base as part of catalysis.[8]
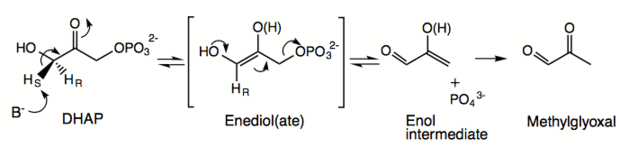
The enzyme first uses Asp71 to specifically abstract the pro-S hydrogen from the C-3 of DHAP to form an ene-diol(ate)-enzyme intermediate, unlike the abstraction of C-3 pro-R hydrogen in TIM by Glu165.[4][8][9] A second base deprotonates the hydroxyl group, leading to the collapse of the en-diol(ate) to form the 2-hydroxy 2-propenal enol intermediate along with dissocation of inorganic phosophate (–OPO3) through the cleavage of a C-O bond rather than an O-P bond.[4][6][7] This deprotonation is catalyzed by either Asp71 or Asp101.[4][9] Protonatation of the methylene group of the enolate is non-stereospecific.[3][8] The reaction products are released sequentially with methylglyoxal leaving before the inorganic phosphate.[7] MGS is responsible for the racemic mixture of lactate in cells; the production of methylglyoxal and its further metabolism yields L-(+)-lactate and D-(-)-lactate, while deletion of the MGS gene leads to observation of optically pure D-(-)-lactate.[10]
Regulation
Binding of phosphate to the enzyme increases its cooperativity via structural changes that open three DHAP-binding sites.[2] At higher concentrations, however, phosphate acts as an competitive allosteric inhibitor to turn off enzymatic activity, suggesting that diversion to methylglyoxal production occurs under conditions of phosphate starvation.[2][3][11] This inhibition is believed to be caused by bound phosphate and formate mimicking the reaction intermediates (enolate and inorganic phosphate).[3] Additionally, phosphate binding causes rotation of threonine residues that close the active site.[3]
Ser55 in the active site of MGS is responsible for discriminating the binding of an inorganic phosphate from the phosphate group of the substrate (DHAP) by hydrogen bonding and undergoing a conformational change of location.[11] Transmittance of the allosteric signal is determined to pass through Arg97 and Val101 because none of these are located in the active site, yet mutations at these residues negates any inhibitory effect of phosphate binding. Pro82 is necessary to transmit the signal from one subunit to the Ar97 and Val101 of another subunit.[11] The induction of salt-bridge formation between Asp10 and Arg140 is an additional inter-subunit signal transmission pathway for organisms that retain the last 10 amino acids of the monomer peptide.[12] The final acceptor of this allosteric signal is the catalytic Gly56 within the active site.[11]
Inorganic pyrophosphate has 95% the ability of phosphate in inhibiting MGS. 3-phosphoglycerate and phosphoenolpyruvate also have 50% and 70% inhibition, respectively.[1] 2-phosphoglycolate also acts as a competitive inhibitor by mimicking the ene-diolate intermediate.[4] ATP has been shown to have weak inhibition in some bacterial strains.[5] The reaction product, methylglyoxal, does not exhibit any feedback inhibition on MGS.[1][6]
Biological Function
Methylglyoxal synthase provides an alternative catabolic pathway for triose phosphates created in glycolysis.[2] It has activity levels similar to that of glyceraldehyde-3-phosphate dehydrogenase from glycolysis, suggesting an interplay between the two enzymes in the breakdown of triose phosphates. Indeed, MGS is strongly inhibited by phosphate concentrations that are close to the Km of phosphate serving as substrate for glyceraldehyde-3-phosphate dehydrogenase and is, therefore, inactive at normal intracellular conditions.[1][2] Triose phosphate catabolism switches over to MGS when phosphate concentrations are too low for glyceraldehyde-3-phosphate dehydrogenase activity.
In situations when glycolysis is restricted by phosphate starvation, the switch to MGS serves to release phosphate from glycolytic metabolites for glyceraldehyde-3-phosphate dehydrogenase and to produce methylglyoxal, which is converted to pyruvate via lactate with the uncoupling of ATP synthesis.[2][8] This interplay between the two enzymes allows the cell to shift triose catabolism between the formation of 1,3-biphosphoglycerate and methylglyoxal based on available phosphates.
Other Applications
For fuel ethanol production, complete metabolism of complex combinations of sugars in E. coli by synthetic biocatalysts is necessary. Deletion of the methylglyoxal synthase gene in E. coli increases fermentation rate of ethanogenic E. coli by promoting the co-metabolism of sugar mixtures containing the five principal sugars found in biomass (glucose, xylose, arabinose, galactose, and mannose).[13] This suggests that MGS production of methylglyoxal plays a role in controlling expression of sugar-specific transporters and catabolic genes in native E.coli.
MGS also has industrial importance in the production of lactate, hydroxyacetone (acetol), and 1,2-propandiol.[5][11][14] Introduction of the MGS gene in bacteria that natively lack MGS increased useful production of 1,2-propandiol by 141%.[14]
For biotech and synthetic applications, phosphate binding helps stabilize and protect the enzyme against cold- and heat-induced denaturation.[7] His-His interaction via the insertion of one histidine residue between Arg22 and His23 is also known to confer greater thermostability by increasing its half-life 4.6-fold.[15]
References
- 1 2 3 4 5 Hopper, D. J.; Cooper, R. A. (1971). "The regulation of Escherichia coli methylglyoxal synthase; a new control site in glycolysis?". FEBS Letters. 13 (4): 213–216. doi:10.1016/0014-5793(71)80538-0. PMID 11945670.
- 1 2 3 4 5 6 7 8 Hopper, D. J.; Cooper, R. A. (1972). "The purification and properties of Escherichia coli methylglyoxal synthase". The Biochemical Journal. 128 (2): 321–9. doi:10.1042/bj1280321. PMC 1173767
 . PMID 4563643.
. PMID 4563643. - 1 2 3 4 5 6 7 8 9 10 11 12 13 Saadat, D; Harrison, D. H. (1999). "The crystal structure of methylglyoxal synthase from Escherichia coli". Structure (London, England : 1993). 7 (3): 309–17. doi:10.1016/s0969-2126(99)80041-0. PMID 10368300.
- 1 2 3 4 5 Saadat, D; Harrison, D. H. (1998). "Identification of catalytic bases in the active site of Escherichia coli methylglyoxal synthase: Cloning, expression, and functional characterization of conserved aspartic acid residues". Biochemistry. 37 (28): 10074–86. doi:10.1021/bi980409p. PMID 9665712.
- 1 2 3 Huang, K; Rudolph, F. B.; Bennett, G. N. (1999). "Characterization of methylglyoxal synthase from Clostridium acetobutylicum ATCC 824 and its use in the formation of 1, 2-propanediol". Applied and Environmental Microbiology. 65 (7): 3244–7. PMC 91483. PMID 10388730.
- 1 2 3 Iyengar, R; Rose, I. A. (1981). "Liberation of the triosephosphate isomerase reaction intermediate and its trapping by isomerase, yeast aldolase, and methylglyoxal synthase". Biochemistry. 20 (5): 1229–35. doi:10.1021/bi00508a027. PMID 7013791.
- 1 2 3 4 Yuan, P. M.; Gracy, R. W. (1977). "The conversion of dihydroxyacetone phosphate to methylglyoxal and inorganic phosphate by methylglyoxal synthase". Archives of Biochemistry and Biophysics. 183 (1): 1–6. doi:10.1016/0003-9861(77)90411-8. PMID 334078.
- 1 2 3 4 Summers, M. C.; Rose, I. A. (1977). "Proton transfer reactions of methylglyoxal synthase". Journal of the American Chemical Society. 99 (13): 4475–8. doi:10.1021/ja00455a044. PMID 325056.
- 1 2 Marks, G. T.; Harris, T. K.; Massiah, M. A.; Mildvan, A. S.; Harrison, D. H. (2001). "Mechanistic implications of methylglyoxal synthase complexed with phosphoglycolohydroxamic acid as observed by X-ray crystallography and NMR spectroscopy". Biochemistry. 40 (23): 6805–18. doi:10.1021/bi0028237. PMID 11389594.
- ↑ Grabar, T. B.; Zhou, S; Shanmugam, K. T.; Yomano, L. P.; Ingram, L. O. (2006). "Methylglyoxal bypass identified as source of chiral contamination in l(+) and d(-)-lactate fermentations by recombinant Escherichia coli". Biotechnology Letters. 28 (19): 1527–35. doi:10.1007/s10529-006-9122-7. PMID 16868860.
- 1 2 3 4 5 Falahati, H; Pazhang, M; Zareian, S; Ghaemi, N; Rofougaran, R; Hofer, A; Rezaie, A. R.; Khajeh, K (2013). "Transmitting the allosteric signal in methylglyoxal synthase". Protein Engineering Design and Selection. 26 (7): 445–52. doi:10.1093/protein/gzt014. PMID 23592737.
- ↑ Zareian, S; Khajeh, K; Pazhang, M; Ranjbar, B (2012). "Rationalization of allosteric pathway in Thermus sp. GH5 methylglyoxal synthase". BMB reports. 45 (12): 748–53. doi:10.5483/bmbrep.2012.45.12.11-138. PMC 4133812. PMID 23261063.
- ↑ Yomano, L. P.; York, S. W.; Shanmugam, K. T.; Ingram, L. O. (2009). "Deletion of methylglyoxal synthase gene (mgsA) increased sugar co-metabolism in ethanol-producing Escherichia coli". Biotechnology Letters. 31 (9): 1389–98. doi:10.1007/s10529-009-0011-8. PMC 2721133. PMID 19458924.
- 1 2 Jung, J. Y.; Yun, H. S.; Lee, J; Oh, M. K. (2011). "Production of 1,2-propanediol from glycerol in Saccharomyces cerevisiae". Journal of microbiology and biotechnology. 21 (8): 846–53. doi:10.4014/jmb.1103.03009. PMID 21876375.
- ↑ Mohammadi, M; Kashi, M. A.; Zareian, S; Mirshahi, M; Khajeh, K (2014). "Remarkable improvement of methylglyoxal synthase thermostability by His-His interaction". Applied Biochemistry and Biotechnology. 172 (1): 157–67. doi:10.1007/s12010-013-0404-y. PMID 24057302.
- Cooper RA, Anderson A (1970). "The formation and catabolism of methylglyoxal during glycolysis in Escherichia coli". FEBS Lett. 11 (4): 273–276. doi:10.1016/0014-5793(70)80546-4. PMID 11945504.
- Hopper DJ, Cooper RA (1971). "The regulation of Escherichia coli methylglyoxal synthase; a new control site in glycolysis?". FEBS Lett. 13 (4): 213–216. doi:10.1016/0014-5793(71)80538-0. PMID 11945670.
- Ray S, Ray M (1981). "Isolation of methylglyoxal synthase from goat liver". J. Biol. Chem. 256 (12): 6230–3. PMID 7240200.
